Working on a remote HPC system
Overview
Teaching: 20 min
Exercises: 0 minQuestions
What is an HPC system?
How does an HPC system work?
How do I log in to a remote HPC system?
Objectives
Connect to a remote HPC system.
Understand the general HPC system architecture.
What Is an HPC System?
The words “cloud”, “cluster”, and the phrase “high-performance computing” or “HPC” are used a lot in different contexts and with various related meanings. So what do they mean? And more importantly, how do we use them in our work?
A Remote computer is one you have no access to physically and must connect via a network (as opposed to Local)
Cloud refers to remote computing resources that are provisioned to users on demand or as needed.
HPC, High Performance Computer, High Performance Computing or Supercomputer are all general terms for a large or powerful computing resource.
Cluster is a more specific term describing a type of supercomputer comprised of multiple smaller computers (nodes) working together. Almost all supercomputers are clusters.

Access
You will connect to a cluster over the internet either with a web client (Jupyter) or with SSH (Secure Shell). Your main interface with the cluster will be using command line.
Nodes
Individual computers that compose a cluster are typically called nodes. On a cluster, there are different types of nodes for different types of tasks. The node where you are now will be different depending on how you accessed the cluster.
Most of you (using JupyterHub) will be on an interactive compute node. This is because Jupyter sessions are launched as a job. If you are using SSH to connect to the cluster, you will be on a login node. Both JupyterHub and SSH login nodes serve as an access point to the cluster.
The real work on a cluster gets done by the compute nodes. Compute nodes come in many shapes and sizes, but generally are dedicated to long or hard tasks that require a lot of computational resources.
What’s in a Node?
A node is similar in makeup to a regular desktop or laptop, composed of CPUs (sometimes also called processors or cores), memory (or RAM), and disk space. Although, where your laptop might have 8 CPUs and 16GB of memory, a compute node will have hundreds of cores and GB of memory.
-
CPUs are a computer’s tool for running programs and calculations.
-
Memory is for short term storage, containing the information currently being operated on by the CPUs.
-
Disk is for long term storage, data stored here is permanent, i.e. still there even if the computer has been restarted. It is common for nodes to connect to a shared, remote disk.

Differences Between Nodes
Many HPC clusters have a variety of nodes optimized for particular workloads. Some nodes may have larger amount of memory, or specialized resources such as Graphical Processing Units (GPUs).
Dedicated Transfer Nodes
If you want to transfer larger amounts of data to or from the cluster, NeSI offers dedicated transfer nodes using the Globus service. More information on using Globus for large data transfer to and from the cluster can be found here: Globus Transfer Service
Key Points
An HPC system is a set of networked machines.
HPC systems typically provide login nodes and a set of compute nodes.
The resources found on independent (compute) nodes can vary in volume and type (amount of RAM, processor architecture, availability of network mounted filesystems, etc.).
Files saved on shared storage are available on all nodes.
The login node is a shared machine: be considerate of other users.
Navigating Files and Directories
Overview
Teaching: 30 min
Exercises: 10 minQuestions
How can I move around the cluster filesystem
How can I see what files and directories I have?
How can I make new files and directories.
Objectives
Create, edit, manipulate and remove files from command line
Translate an absolute path into a relative path and vice versa.
Use options and arguments to change the behaviour of a shell command.
Demonstrate the use of tab completion and explain its advantages.
The Unix Shell
This episode will be a quick introduction to the Unix shell, only the bare minimum required to use the cluster.
The Software Carpentry ‘Unix Shell’ lesson covers the subject in more depth, we recommend you check it out.
The part of the operating system responsible for managing files and directories is called the file system. It organizes our data into files, which hold information, and directories (also called ‘folders’), which hold files or other directories.
Understanding how to navigate the file system using command line is essential for using an HPC.
The NeSI filesystem looks something like this:

The directories that are relevant to us are.
| Location | Default Storage | Backup | |
| Home is for user-specific files such as configuration files, environment setup, source code, etc. | /home/<username> |
20GB | Daily |
| Project is for persistent project-related data, project-related software, etc. | /nesi/project/<projectcode> |
100GB | Daily |
| Nobackup is a 'scratch space', for data you don't need to keep long term. Old data is periodically deleted from nobackup | /nesi/nobackup/<projectcode> |
10TB | None |
Managing your data and storage (backups and quotas)
NeSI performs backups of the /home and /nesi/project (persistent) filesystems. However, backups are only captured once per day. So, if you edit or change code or data and then immediately delete it, it likely cannot be recovered. Note, as the name suggests, NeSI does not backup the /nesi/nobackup filesystem.
Protecting critical data from corruption or deletion is primarily your
responsibility. Ensure you have a data management plan and stick to the plan to reduce the chance of data loss. Also important is managing your storage quota. To check your quotas, use the nn_storage_quota command, eg
$ nn_storage_quota
Quota Location Available Used Use%
home_johndoe 20G 14.51G 72
project_nesi99991 100G 101G 101
nobackup_nesi99991 10T 0 0
Notice that the project space for this user is over quota and has been locked, meaning no more data can be added. When your space is locked you will need to move or remove data. Also note that none of the nobackup space is being used. Likely data from project can be moved to nobackup. nn_storage_quota uses cached data, and so will no immediately show changes to storage use.
For more details on our persistent and nobackup storage systems, including data retention and the nobackup autodelete schedule, please see our Filesystem and Quota documentation.
Directories are like places — at any time while we are using the shell, we are in exactly one place called our current working directory. Commands mostly read and write files in the current working directory, i.e. ‘here’, so knowing where you are before running a command is important.
First, let’s find out where we are by running the command pwd for ‘print working directory’.
10:00:00 login01 $ pwd
/home/<username>
The output we see is what is known as a ‘path’. The path can be thought of as a series of directions given to navigate the file system.
At the top is the root directory that holds all the files in a filesystem.
We refer to it using a slash character, /, on its own.
This is what the leading slash in /home/<username> is referring to, it is telling us our path starts at the root directory.
Next is home, as it is the next part of the path we know it is inside the root directory,
we also know that home is another directory as the path continues.
Finally, stored inside home is the directory with your username.
Slashes
Notice that there are two meanings for the
/character. When it appears at the front of a file or directory name, it refers to the root directory. When it appears inside a path, it’s just a separator.
As you may now see, using a bash shell is strongly dependent on the idea that your files are organized in a hierarchical file system. Organizing things hierarchically in this way helps us keep track of our work: it’s possible to put hundreds of files in our home directory, just as it’s possible to pile hundreds of printed papers on our desk, but it’s a self-defeating strategy.
Listing the contents of directories
To list the contents of a directory, we use the command ls followed by the path to the directory whose contents we want listed.
We will now list the contents of the directory we we will be working from. We can use the following command to do this:
10:00:00 login01 $ ls /nesi/project/nesi99991
introhpc2510
You should see a directory called introhpc2510, and possibly several other directories. For the purposes of this workshop you will be working within /nesi/project/nesi99991/introhpc2510
Command History
You can cycle through your previous commands with the ↑ and ↓ keys. A convenient way to repeat your last command is to type ↑ then enter.
lsReading ComprehensionWhat command would you type to get the following output
original pnas_final pnas_sub
ls pwdls backupls /Users/backupls /backupSolution
- No:
pwdis not the name of a directory.- Possibly: It depends on your current directory (we will explore this more shortly).
- Yes: uses the absolute path explicitly.
- No: There is no such directory.

Moving about
Currently we are still in our home directory, we want to move into theproject directory from the previous command.
The command to change directory is cd followed by the path to the directory we want to move to.
The cd command is akin to double clicking a folder in a graphical interface.
We will use the following command:
10:00:00 login01 $ cd /nesi/project/nesi99991/introhpc2510
You will notice that cd doesn’t print anything. This is normal. Many shell commands will not output anything to the screen when successfully executed.
We can check we are in the right place by running pwd.
10:00:00 login01 $ pwd
/nesi/project/nesi99991/introhpc2510
Creating directories
As previously mentioned, it is general useful to organise your work in a hierarchical file structure to make managing and finding files easier. It is also is especially important when working within a shared directory with colleagues, such as a project, to minimise the chance of accidentally affecting your colleagues work. So for this workshop you will each make a directory using the mkdir command within the workshops directory for you to personally work from.
10:00:00 login01 $ mkdir <username>
You should then be able to see your new directory is there using ls.
10:00:00 login01 $ ls /nesi/project/nesi99991/introhpc2510
sum_matrix.r usr123 usr345
General Syntax of a Shell Command
We are now going to use ls again but with a twist, this time we will also use what are known as options, flags or switches.
These options modify the way that the command works, for this example we will add the flag -l for “long listing format”.
10:00:00 login01 $ ls -l /nesi/project/nesi99991/introhpc2510
-rw-r-----+ 1 usr9999 nesi99991 460 Nov 18 17:03
-rw-r-----+ 1 usr9999 nesi99991 460 Nov 18 17:03 sum_matrix.r
drwxr-sr-x+ 3 usr9999 nesi99991 4096 22 Sep 08:40 birds
drwxrws---+ 2 usr123 nesi99991 4096 Nov 15 09:01 usr123
drwxrws---+ 2 usr345 nesi99991 4096 Nov 15 09:01 usr345
We can see that the -l option has modified the command and now our output has listed all the files in alphanumeric order, which can make finding a specific file easier.
It also includes information about the file size, time of its last modification, and permission and ownership information.
Most unix commands follow this basic structure.

The prompt tells us that the terminal is accepting inputs, prompts can be customised to show all sorts of info.
The command, what are we trying to do.
Options will modify the behavior of the command, multiple options can be specified.
Options will either start with a single dash (-) or two dashes (--)..
Often options will have a short and long format e.g. -a and --all.
Arguments tell the command what to operate on (usually files and directories).
Each part is separated by spaces: if you omit the space
between ls and -l the shell will look for a command called ls-l, which
doesn’t exist. Also, capitalization can be important.
For example, ls -s will display the size of files and directories alongside the names,
while ls -S will sort the files and directories by size.
Another useful option for ls is the -a option, lets try using this option together with the -l option:
10:00:00 login01 $ ls -la
drwxrws---+ 4 usr001 nesi99991 4096 Nov 15 09:00 .
drwxrws---+ 12 root nesi99991 262144 Nov 15 09:23 ..
-rw-r-----+ 1 cwal219 nesi99991 460 Nov 18 17:03 sum_matrix.r
drwxrws---+ 2 usr123 nesi99991 4096 Nov 15 09:01 usr123
drwxrws---+ 2 usr345 nesi99991 4096 Nov 15 09:01 usr345
Single letter options don’t usually need to be separate. In this case ls -la is performing the same function as if we had typed ls -l -a.
You might notice that we now have two extra lines for directories . and ... These are hidden directories which the -a option has been used to reveal, you can make any file or directory hidden by beginning their filenames with a ..
These two specific hidden directories are special as they will exist hidden inside every directory, with the . hidden directory representing your current directory and the .. hidden directory representing the parent directory above your current directory.
Exploring More
lsFlagsYou can also use two options at the same time. What does the command
lsdo when used with the-loption? What about if you use both the-land the-hoption?Some of its output is about properties that we do not cover in this lesson (such as file permissions and ownership), but the rest should be useful nevertheless.
Solution
The
-loption makeslsuse a long listing format, showing not only the file/directory names but also additional information, such as the file size and the time of its last modification. If you use both the-hoption and the-loption, this makes the file size ‘human readable’, i.e. displaying something like5.3Kinstead of5369.
Relative paths
You may have noticed in the last command we did not specify an argument for the directory path. Until now, when specifying directory names, or even a directory path (as above), we have been using what are known as absolute paths, which work no matter where you are currently located on the machine since it specifies the full path from the top level root directory.
An absolute path always starts at the root directory, which is indicated by a
leading slash. The leading / tells the computer to follow the path from
the root of the file system, so it always refers to exactly one directory,
no matter where we are when we run the command.
Any path without a leading / is a relative path.
When you use a relative path with a command
like ls or cd, it tries to find that location starting from where we are,
rather than from the root of the file system.
In the previous command, since we did not specify an absolute path it ran the command on the relative path from our current directory
(implicitly using the . hidden directory), and so listed the contents of our current directory.
We will now navigate to the parent directory, the simplest way do this is to use the relative path ...
10:00:00 login01 $ cd ..
We should now be back in /nesi/project/nesi99991.
10:00:00 login01 $ pwd
/nesi/project/nesi99991
Tab completion
Sometimes file paths and file names can be very long, making typing out the path tedious. One trick you can use to save yourself time is to use something called tab completion. If you start typing the path in a command and there is only one possible match, if you hit tab the path will autocomplete (until there are more than one possible matches).
For example, if you type:
10:00:00 login01 $ cd int
and then press Tab (the tab key on your keyboard), the shell automatically completes the directory name for you (since there is only one possible match):
10:00:00 login01 $ cd introhpc2510/
However, you want to move to your personal working directory. If you hit Tab once you will likely see nothing change, as there are more than one possible options. Hitting Tab a second time will print all possible autocomplete options.
cwal219/ riom/ harrellw/
Now entering in the first few characters of the path (just enough that the possible options are no longer ambiguous) and pressing Tab again, should complete the path.
Now press Enter to execute the command.
10:00:00 login01 $ cd introhpc2510/<username>
Check that we’ve moved to the right place by running pwd.
/nesi/project/nesi99991/introhpc2510/<username>
Two More Shortcuts
The shell interprets a tilde (
~) character at the start of a path to mean “the current user’s home directory”. For example, if Nelle’s home directory is/home/nelle, then~/datais equivalent to/home/nelle/data. This only works if it is the first character in the path:here/there/~/elsewhereis nothere/there//home/nelle/elsewhere.Another shortcut is the
-(dash) character.cdwill translate-into the previous directory I was in, which is faster than having to remember, then type, the full path. This is a very efficient way of moving back and forth between two directories – i.e. if you executecd -twice, you end up back in the starting directory.The difference between
cd ..andcd -is that the former brings you up, while the latter brings you back.
Absolute vs Relative Paths
Starting from
/home/amanda/data, which of the following commands could Amanda use to navigate to her home directory, which is/home/amanda?
cd .cd /cd home/amandacd ../..cd ~cd homecd ~/data/..cdcd ..Solution
- No:
.stands for the current directory.- No:
/stands for the root directory.- No: Amanda’s home directory is
/home/amanda.- No: this command goes up two levels, i.e. ends in
/home.- Yes:
~stands for the user’s home directory, in this case/home/amanda.- No: this command would navigate into a directory
homein the current directory if it exists.- Yes: unnecessarily complicated, but correct.
- Yes: shortcut to go back to the user’s home directory.
- Yes: goes up one level.
Relative Path Resolution
Using the filesystem diagram below, if
pwddisplays/Users/thing, what willls ../backupdisplay?
../backup: No such file or directory2012-12-01 2013-01-08 2013-01-27original pnas_final pnas_sub
Solution
- No: there is a directory
backupin/Users.- No: this is the content of
Users/thing/backup, but with.., we asked for one level further up.- Yes:
../backup/refers to/Users/backup/.
Clearing your terminal
If your screen gets too cluttered, you can clear your terminal using the
clearcommand. You can still access previous commands using ↑ and ↓ to move line-by-line, or by scrolling in your terminal.
Listing in Reverse Chronological Order
By default,
lslists the contents of a directory in alphabetical order by name. The commandls -tlists items by time of last change instead of alphabetically. The commandls -rlists the contents of a directory in reverse order. Which file is displayed last when you combine the-tand-rflags? Hint: You may need to use the-lflag to see the last changed dates.Solution
The most recently changed file is listed last when using
-rt. This can be very useful for finding your most recent edits or checking to see if a new output file was written.
Globbing
One of the most powerful features of bash is filename expansion, otherwise known as globbing. This allows you to use patterns to match a file name (or multiple files), which will then be operated on as if you had typed out all of the matches.
*is a wildcard, which matches zero or more characters.Inside the
/nesi/project/nesi99991/introhpc2510directory there is a directory calledbirds10:00:00 login01 $ cd /nesi/project/nesi99991/introhpc2510/birds 10:00:00 login01 $ lskaka.txt kakapo.jpeg kea.txt kiwi.jpeg pukeko.jpegIn this example there aren’t many files, but it is easy to imagine a situation where you have hundreds or thousads of files you need to filter through, and globbing is the perfect tool for this. Using the wildcard character the command
10:00:00 login01 $ ls ka*Will return:
kaka.txt kakapo.jpegSince the pattern
ka*will matchkaka.txtandkakapo.jpegas these both start with “ka”. While the command:10:00:00 login01 $ ls *.jpegWill return:
kakapo.jpeg kiwi.jpeg pukeko.jpegAs
*.jpegwill matchkakapo.jpeg,kiwi.jpegandpukeko.jpegas they all end in.jpegYou can use multiple wildcards as well with the command:10:00:00 login01 $ ls k*a.*Returning:
kaka.txt kea.txtAs
k*a.*will match justkaka.txtandkea.txt
?is also a wildcard, but it matches exactly one character. So the command:10:00:00 login01 $ ls ????.*Would return:
kaka.txt kiwi.jpegAs
kaka.txtandkiwi.jpegthe only files which have four characters, followed by a.then any number and combination of characters.When the shell sees a wildcard, it expands the wildcard to create a list of matching filenames before running the command that was asked for. As an exception, if a wildcard expression does not match any file, Bash will pass the expression as an argument to the command as it is. However, generally commands like
wcandlssee the lists of file names matching these expressions, but not the wildcards themselves. It is the shell, not the other programs, that deals with expanding wildcards.
List filenames matching a pattern
Running
lsin a directory gives the outputcubane.pdb ethane.pdb methane.pdb octane.pdb pentane.pdb propane.pdbWhich
lscommand(s) will produce this output?
ethane.pdb methane.pdb
ls *t*ane.pdbls *t?ne.*ls *t??ne.pdbls ethane.*Solution
The solution is
3.
1.shows all files whose names contain zero or more characters (*) followed by the lettert, then zero or more characters (*) followed byane.pdb. This givesethane.pdb methane.pdb octane.pdb pentane.pdb.
2.shows all files whose names start with zero or more characters (*) followed by the lettert, then a single character (?), thenne.followed by zero or more characters (*). This will give usoctane.pdbandpentane.pdbbut doesn’t match anything which ends inthane.pdb.
3.fixes the problems of option 2 by matching two characters (??) betweentandne. This is the solution.
4.only shows files starting withethane..
include in terminal excersise (delete slurm files later on maybe?)
Create a text file
Now let’s create a file. To do this we will use a text editor called Nano to create a file called draft.txt:
10:00:00 login01 $ nano draft.txt
Which Editor?
When we say, ‘
nanois a text editor’ we really do mean ‘text’: it can only work with plain character data, not tables, images, or any other human-friendly media. We use it in examples because it is one of the least complex text editors. However, because of this trait, it may not be powerful enough or flexible enough for the work you need to do after this workshop. On Unix systems (such as Linux and macOS), many programmers use Emacs or Vim (both of which require more time to learn), or a graphical editor such as Gedit. On Windows, you may wish to use Notepad++. Windows also has a built-in editor callednotepadthat can be run from the command line in the same way asnanofor the purposes of this lesson.No matter what editor you use, you will need to know where it searches for and saves files. If you start it from the shell, it will (probably) use your current working directory as its default location. If you use your computer’s start menu, it may want to save files in your desktop or documents directory instead. You can change this by navigating to another directory the first time you ‘Save As…’
Let’s type in a few lines of text.
Once we’re happy with our text, we can press Ctrl+O
(press the Ctrl or Control key and, while
holding it down, press the O key) to write our data to disk
(we’ll be asked what file we want to save this to:
press Return to accept the suggested default of draft.txt).
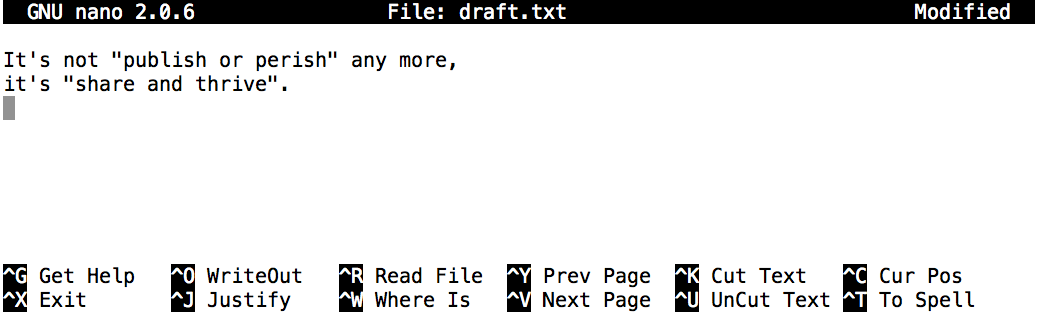
Once our file is saved, we can use Ctrl+X to quit the editor and return to the shell.
Control, Ctrl, or ^ Key
The Control key is also called the ‘Ctrl’ key. There are various ways in which using the Control key may be described. For example, you may see an instruction to press the Control key and, while holding it down, press the X key, described as any of:
Control-XControl+XCtrl-XCtrl+X^XC-xIn nano, along the bottom of the screen you’ll see
^G Get Help ^O WriteOut. This means that you can useControl-Gto get help andControl-Oto save your file.
nano doesn’t leave any output on the screen after it exits,
but ls now shows that we have created a file called draft.txt:
10:00:00 login01 $ ls
draft.txt
Copying files and directories
In a future lesson, we will be running the R script /nesi/project/nesi99991/introhpc2510/sum_matrix.r, but as we can’t all work on the same file at once you will need to take your own copy. This can be done with the copy command cp, at least two arguments are needed the file (or directory) you want to copy, and the directory (or file) where you want the copy to be created. We will be copying the file into the directory we made previously, as this should be your current directory the second argument can be a simple ..
10:00:00 login01 $ cp /nesi/project/nesi99991/introhpc2510/sum_matrix.r .
We can check that it did the right thing using ls
10:00:00 login01 $ ls
draft.txt sum_matrix.r
Other File operations
cat stands for concatenate, meaning to link or merge things together. It is primarily used for printing the contents of one or more files to the standard output.
head and tail will print the first or last lines (head or tail) of the specified file(s). By default it will print 10 lines, but a specific number of lines can be specified with the -n option.
mv to move move a file, is used similarly to cp taking a source argument(s) and a destination argument.
rm will remove move a file and only needs one argument.
The mv command is also used to rename a file, for example mv my_fiel my_file. This is because as far as the computer is concerned moving and renaming a file are the same operation.
In order to cp a directory (and all its contents) the -r for recursive option must be used.
The same is true when deleting directories with rm
| command | name | usage |
|---|---|---|
cp |
copy | cp file1 file2 |
cp -r directory1/ directory2/ |
||
mv |
move | mv file1 file2 |
mv directory1/ directory2/ |
||
rm |
remove | rm file1 file2 |
rm -r directory1/ directory2/ |
||
cat |
concatinate | cat file1 |
cat file1 file2 |
||
head |
head | head file1 |
head -n 5 file1 |
||
tail |
tail | tail file1 |
tail -n 5 file1 |
For mv and cp if the destination path (final argument) is an existing directory the file will be placed inside that directory with the same name as the source.
Moving vs Copying
When using the
cporrmcommands on a directory the ‘recursive’ flag-rmust be used, butmvdoes not require it?Solution
We mentioned previously that as far the computer is concerned, renaming is the same operation as moving. Contrary to what the commands name implies, all moving is actually renaming. The data on the hard drive stays in the same place, only the label applied to that block of memory is changed. To copy a directory, each individual file inside that directory must be read, and then written to the copy destination. To delete a directory, each individual file in the directory must be marked for deletion, however when moving a directory the files inside are the data inside the directory is not interacted with, only the parent directory is “renamed” to a different place.
This is also why
mvis faster thancpas no reading of the files is required.
Unsupported command-line options
If you try to use an option (flag) that is not supported,
lsand other commands will usually print an error message similar to:$ ls -jls: invalid option -- 'j' Try 'ls --help' for more information.
Getting help
Commands will often have many options. Most commands have a --help flag, as can be seen in the error above. You can also use the manual pages (aka manpages) by using the man command. The manual page provides you with all the available options and their use in more detail. For example, for thr ls command:
10:00:00 login01 $ man ls
Usage: ls [OPTION]... [FILE]...
List information about the FILEs (the current directory by default).
Sort entries alphabetically if neither -cftuvSUX nor --sort is specified.
Mandatory arguments to long options are mandatory for short options, too.
-a, --all do not ignore entries starting with .
-A, --almost-all do not list implied . and ..
--author with -l, print the author of each file
-b, --escape print C-style escapes for nongraphic characters
--block-size=SIZE scale sizes by SIZE before printing them; e.g.,
'--block-size=M' prints sizes in units of
1,048,576 bytes; see SIZE format below
-B, --ignore-backups do not list implied entries ending with ~
-c with -lt: sort by, and show, ctime (time of last
modification of file status information);
with -l: show ctime and sort by name;
otherwise: sort by ctime, newest first
-C list entries by columns
--color[=WHEN] colorize the output; WHEN can be 'always' (default
if omitted), 'auto', or 'never'; more info below
-d, --directory list directories themselves, not their contents
-D, --dired generate output designed for Emacs' dired mode
-f do not sort, enable -aU, disable -ls --color
-F, --classify append indicator (one of */=>@|) to entries
... ... ...
To navigate through the man pages,
you may use ↑ and ↓ to move line-by-line,
or try B and Spacebar to skip up and down by a full page.
To search for a character or word in the man pages,
use / followed by the character or word you are searching for.
Sometimes a search will result in multiple hits. If so, you can move between hits using N (for moving forward) and Shift+N (for moving backward).
To quit the man pages, press Q.
Manual pages on the web
Of course, there is a third way to access help for commands: searching the internet via your web browser. When using internet search, including the phrase
unix man pagein your search query will help to find relevant results.GNU provides links to its manuals including the core GNU utilities, which covers many commands introduced within this lesson.
Key Points
The file system is responsible for managing information on the disk.
Information is stored in files, which are stored in directories (folders).
Directories can also store other directories, which then form a directory tree.
cd [path]changes the current working directory.
ls [path]prints a listing of a specific file or directory;lson its own lists the current working directory.
pwdprints the user’s current working directory.
cp [file] [path]copies [file] to [path]
mv [file] [path]moves [file] to [path]
rm [file]deletes [file]
/on its own is the root directory of the whole file system.Most commands take options (flags) that begin with a
-.A relative path specifies a location starting from the current location.
An absolute path specifies a location from the root of the file system.
Directory names in a path are separated with
/on Unix, but\on Windows.
..means ‘the directory above the current one’;.on its own means ‘the current directory’.
Morning Break
Overview
Teaching: min
Exercises: minQuestions
Objectives
Key Points
Accessing software via Modules
Overview
Teaching: 20 min
Exercises: 5 minQuestions
How do we load and unload software packages?
Objectives
Load and use a software package.
Explain how the shell environment changes when the module mechanism loads or unloads packages.
On a high-performance computing system, it is seldom the case that the software we want to use is available when we log in. It is installed, but we will need to “load” it before it can run.
Before we start using individual software packages, however, we should understand the reasoning behind this approach. The three biggest factors are:
- software incompatibilities
- versioning
- dependencies
Software incompatibility is a major headache for programmers. Sometimes the
presence (or absence) of a software package will break others that depend on
it. Two of the most famous examples are Python 2 and 3 and C compiler versions.
Python 3 famously provides a python command that conflicts with that provided
by Python 2. Software compiled against a newer version of the C libraries and
then used when they are not present will result in a nasty 'GLIBCXX_3.4.20'
not found error, for instance.
Software versioning is another common issue. A team might depend on a certain package version for their research project - if the software version was to change (for instance, if a package was updated), it might affect their results. Having access to multiple software versions allows a set of researchers to prevent software versioning issues from affecting their results.
Dependencies are where a particular software package (or even a particular version) depends on having access to another software package (or even a particular version of another software package). For example, the VASP materials science software may depend on having a particular version of the FFTW (Fastest Fourier Transform in the West) software library available for it to work.
Environment
Before understanding environment modules we first need to understand what is meant by environment.
The environment is defined by it’s environment variables.
Environment Variables are writable named-variables.
We can assign a variable named “FOO” with the value “bar” using the syntax.
10:00:00 login01 $ FOO="bar"
Convention is to name fixed variables in all caps.
Our new variable can be referenced using $FOO, you could also use ${FOO},
enclosing a variable in curly brackets is good practice as it avoids possible ambiguity.
10:00:00 login01 $ $FOO
-bash: bar: command not found
We got an error here because the variable is evalued in the terminal then executed. If we just want to print the variable we can use the command,
10:00:00 login01 $ echo $FOO
bar
We can get a full list of environment variables using the command,
10:00:00 login01 $ env
[removed some lines for clarity]
EBROOTTCL=/opt/nesi/CS400_centos7_bdw/Tcl/8.6.10-GCCcore-9.2.0
CPUARCH_STRING=bdw
TERM=xterm-256color
SHELL=/bin/bash
EBROOTGCCCORE=/opt/nesi/CS400_centos7_bdw/GCCcore/9.2.0
EBDEVELFREETYPE=/opt/nesi/CS400_centos7_bdw/freetype/2.10.1-GCCcore-9.2.0/easybuild/freetype-2.10.1-GCCcore-9.2.0-easybuild-devel
HISTSIZE=10000
XALT_EXECUTABLE_TRACKING=yes
MODULEPATH_ROOT=/usr/share/modulefiles
LMOD_SYSTEM_DEFAULT_MODULES=NeSI
SSH_CLIENT=192.168.94.65 45946 22
EBDEVELMETIS=/opt/nesi/CS400_centos7_bdw/METIS/5.1.0-GCCcore-9.2.0/easybuild/METIS-5.1.0-GCCcore-9.2.0-easybuild-devel
XALT_DIR=/opt/nesi/CS400_centos7_bdw/XALT/current
LMOD_PACKAGE_PATH=/opt/nesi/share/etc/lmod
These variables control many aspects of how your terminal, and any software launched from your terminal works.
Environment Modules
Environment modules are the solution to these problems. A module is a self-contained description of a software package – it contains the settings required to run a software package and, usually, encodes required dependencies on other software packages.
There are a number of different environment module implementations commonly
used on HPC systems: the two most common are TCL modules and Lmod. Both of
these use similar syntax and the concepts are the same so learning to use one
will allow you to use whichever is installed on the system you are using. In
both implementations the module command is used to interact with environment
modules. An additional subcommand is usually added to the command to specify
what you want to do. For a list of subcommands you can use module -h or
module help. As for all commands, you can access the full help on the man
pages with man module.
Purging Modules
Depending on how you are accessing the HPC the modules you have loaded by default will be different. So before we start listing our modules we will first use the module purge command to clear all but the minimum default modules so that we are all starting with the same modules.
10:00:00 login01 $ module purge
The following modules were not unloaded:
(Use "module --force purge" to unload all):
1) NeSI/zen3
Note that module purge is informative. It lets us know that all but a minimal default
set of packages have been unloaded (and how to actually unload these if we
truly so desired).
We are able to unload individual modules, unfortunately within the NeSI system it does not always unload it’s dependencies, therefore we recommend module purge to bring you back to a state where only those modules needed to perform your normal work on the cluster.
module purge is a useful tool for ensuring repeatable research by guaranteeing that the environment that you build your software stack from is always the same. This is important since some modules have the potential to silently effect your results if they are loaded (or not loaded).
Listing Available Modules
To see available software modules, use module avail:
10:00:00 login01 $ module avail
-----------------/opt/nesi/CS400_centos7_bdw/modules/all ------------------
fgbio/2.0.2 PyQt/5.10.1-gimkl-2018b-Python-3.7.3
FIGARO/1.1.2-gimkl-2022a-Python-3.11.3 PyQt/5.12.1-gimkl-2020a-Python-3.8.2 (D)
File-Rename/1.13-GCC-9.2.0 Python-Geo/2.7.16-gimkl-2018b
FileSender/2.49.0-foss-2023a Python-Geo/3.7.3-gimkl-2018b
Filtlong/0.2.0 Python-Geo/3.8.2-gimkl-2020a
FimTyper/1.0.1-GCC-11.3.0-Perl-5.34.1 Python-Geo/3.9.5-gimkl-2020a
fineSTRUCTURE/4.1.1-gimkl-2022a-Perl-5.34.1 Python-Geo/3.9.9-gimkl-2020a
flatbuffers/1.12.0-GCCcore-9.2.0 Python-Geo/3.10.5-gimkl-2022a
flex/2.6.4-GCCcore-7.4.0 Python-Geo/3.11.3-gimkl-2022a (D)
FlexiBLAS/3.1.3-GCC-11.3.0 Python/2.7.16-gimkl-2018b
FlexiBLAS/3.3.1-GCC-12.3.0 Python/2.7.18-gimkl-2020a
FlexiBLAS/3.4.4-intel-compilers-2023.2.1 (D) Python/3.7.3-gimkl-2018b
FLTK/1.3.5-GCCcore-9.2.0 Python/3.8.1-gimkl-2018b
Flye/2.4.2-gimkl-2018b-Python-2.7.16 Python/3.8.2-gimkl-2020a
Flye/2.7.1-gimkl-2020a-Python-2.7.18 Python/3.9.5-gimkl-2020a
Flye/2.8.3-gimkl-2020a-Python-3.8.2 Python/3.9.9-gimkl-2020a
Flye/2.9-gimkl-2020a-Python-3.8.2 Python/3.10.5-gimkl-2022a
Flye/2.9.1-gimkl-2022a-Python-3.10.5 Python/3.11.3-gimkl-2022a
Flye/2.9.3-gimkl-2022a-Python-3.11.3 Python/3.11.6-foss-2023a (D)
[removed most of the output here for clarity]
Where:
S: Module is Sticky, requires --force to unload or purge
L: Module is loaded
D: Default Module
If the avail list is too long consider trying:
"module --default avail" or "ml -d av" to just list the default modules.
"module overview" or "ml ov" to display the number of modules for each name.
Use "module spider" to find all possible modules and extensions.
Use "module keyword key1 key2 ..." to search for all possible modules matching any of the "keys".
Listing Currently Loaded Modules
You can use the module list command to see which modules you currently have
loaded in your environment. On mahuika you will have a few default modules loaded when you login.
10:00:00 login01 $ module list
Currently Loaded Modules:
1) NeSI/zen3 (S)
Where:
S: Module is Sticky, requires --force to unload or purge
Loading and Unloading Software
You can load software using the module load command. In this example we will be using the programming language R.
Initially, R is not loaded. We can test this by using the which
command. which looks for programs the same way that Bash does, so we can use
it to tell us where a particular piece of software is stored.
10:00:00 login01 $ which R
/usr/bin/which: no R in (/usr/local/bin:/usr/bin:/usr/local/sbin:/usr/sbin:/opt/nesi/bin)
The important bit here being:
/usr/bin/which: no R in (...)
Now lets try loading the R environment module, and try again.
10:00:00 login01 $ module load R
10:00:00 login01 $ which R
/opt/nesi/zen3/R/4.3.2-foss-2023a/bin/R
Tab Completion
The module command also supports tab completion. You may find this the easiest way to find the right software.
So, what just happened?
To understand the output, first we need to understand the nature of the $PATH
environment variable. $PATH is a special environment variable that controls
where a UNIX system looks for software. Specifically $PATH is a list of
directories (separated by :) that the OS searches through for a command
before giving up and telling us it can’t find it. As with all environment
variables we can print it out using echo.
10:00:00 login01 $ echo $PATH
/opt/nesi/zen3/R/4.3.2-foss-2023a/bin:/opt/nesi/CS400_centos7_bdw/LibTIFF/4.4.0-GCCcore-12.3.0/bin:/opt/nesi/CS400_centos7_bdw/nodejs/18.18.2-GCCcore-12.3.0/bin:/opt/nesi/CS400_centos7_bdw/libgit2/1.6.4-GCC-12.3.0/bin:/opt/nesi/CS400_centos7_bdw/Java/20.0.2:/opt/nesi/CS400_centos7_bdw/Java/20.0.2/bin:/opt/nesi/CS400_centos7_bdw/cURL/8.3.0-GCCcore-12.3.0/bin:/opt/nesi/CS400_centos7_bdw/OpenSSL/1.1/bin:/opt/nesi/CS400_centos7_bdw/SQLite/3.42.0-GCCcore-12.3.0/bin:/opt/nesi/CS400_centos7_bdw/libxml2/2.11.4-GCCcore-12.3.0/bin:/opt/nesi/CS400_centos7_bdw/ncurses/6.4-GCCcore-12.3.0/bin:/opt/nesi/CS400_centos7_bdw/PCRE2/10.42-GCCcore-12.3.0/bin:/opt/nesi/zen3/XZ/5.4.2-GCCcore-12.3.0/bin:/opt/nesi/CS400_centos7_bdw/bzip2/1.0.8-GCCcore-12.3.0/bin:/opt/nesi/CS400_centos7_bdw/FFTW/3.3.10-GCC-12.3.0/bin:/opt/nesi/CS400_centos7_bdw/FlexiBLAS/3.3.1-GCC-12.3.0/bin:/opt/nesi/zen3/OpenMPI/4.1.5-GCC-12.3.0/bin:/opt/nesi/zen3/UCC/1.3.0-GCCcore-12.3.0/bin:/opt/nesi/zen3/UCX/1.18.1-GCCcore-12.3.0/bin:/opt/nesi/CS400_centos7_bdw/numactl/2.0.16-GCCcore-12.3.0/bin:/opt/nesi/CS400_centos7_bdw/binutils/2.40-GCCcore-12.3.0/bin:/opt/nesi/CS400_centos7_bdw/GCCcore/12.3.0/bin:/usr/local/bin:/usr/bin:/usr/local/sbin:/usr/sbin:/opt/nesi/bin
We can improve the readability of this command slightly by replacing the colon delimiters (:) with newline (\n) characters.
10:00:00 login01 $ echo $PATH | tr ":" "\n"
/opt/nesi/zen3/R/4.3.2-foss-2023a/bin
/opt/nesi/CS400_centos7_bdw/LibTIFF/4.4.0-GCCcore-12.3.0/bin
/opt/nesi/CS400_centos7_bdw/nodejs/18.18.2-GCCcore-12.3.0/bin
/opt/nesi/CS400_centos7_bdw/libgit2/1.6.4-GCC-12.3.0/bin
/opt/nesi/CS400_centos7_bdw/Java/20.0.2
/opt/nesi/CS400_centos7_bdw/Java/20.0.2/bin
/opt/nesi/CS400_centos7_bdw/cURL/8.3.0-GCCcore-12.3.0/bin
/opt/nesi/CS400_centos7_bdw/OpenSSL/1.1/bin
/opt/nesi/CS400_centos7_bdw/SQLite/3.42.0-GCCcore-12.3.0/bin
/opt/nesi/CS400_centos7_bdw/libxml2/2.11.4-GCCcore-12.3.0/bin
/opt/nesi/CS400_centos7_bdw/ncurses/6.4-GCCcore-12.3.0/bin
/opt/nesi/CS400_centos7_bdw/PCRE2/10.42-GCCcore-12.3.0/bin
/opt/nesi/zen3/XZ/5.4.2-GCCcore-12.3.0/bin
/opt/nesi/CS400_centos7_bdw/bzip2/1.0.8-GCCcore-12.3.0/bin
/opt/nesi/CS400_centos7_bdw/FFTW/3.3.10-GCC-12.3.0/bin
/opt/nesi/CS400_centos7_bdw/FlexiBLAS/3.3.1-GCC-12.3.0/bin
/opt/nesi/zen3/OpenMPI/4.1.5-GCC-12.3.0/bin
/opt/nesi/zen3/UCC/1.3.0-GCCcore-12.3.0/bin
/opt/nesi/zen3/UCX/1.18.1-GCCcore-12.3.0/bin
/opt/nesi/CS400_centos7_bdw/numactl/2.0.16-GCCcore-12.3.0/bin
/opt/nesi/CS400_centos7_bdw/binutils/2.40-GCCcore-12.3.0/bin
/opt/nesi/CS400_centos7_bdw/GCCcore/12.3.0/bin
/usr/local/bin
/usr/bin
/usr/local/sbin
/usr/sbin
/opt/nesi/bin
You’ll notice a similarity to the output of the which command. However, in this case,
there are a lot more directories at the beginning. When we
ran the module load command, it added many directories to the beginning of our
$PATH.
You may have noticed that the first directory to appear is /opt/nesi/zen3/R/4.3.2-foss-2023a/bin. Let’s examine what’s there:
10:00:00 login01 $ ls /opt/nesi/zen3/R/4.3.2-foss-2023a/bin
R Rscript
module load “loads” not only the specified software, but it also loads software dependencies. That is, the software that the application you load requires to run.
To demonstrate, let’s use module list.
10:00:00 login01 $ module list
1) NeSI/zen3 (S) 12) FFTW/3.3.10-GCC-12.3.0 23) SQLite/3.42.0-GCCcore-12.3.0
2) GCCcore/12.3.0 13) gompi/2023a 24) OpenSSL/1.1
3) zlib/1.2.13-GCCcore-12.3.0 14) FFTW.MPI/3.3.10-gompi-2023a 25) cURL/8.3.0-GCCcore-12.3.0
4) binutils/2.40-GCCcore-12.3.0 15) ScaLAPACK/2.2.0-gompi-2023a-fb 26) NLopt/2.7.1-GCC-12.3.0
5) GCC/12.3.0 16) foss/2023a 27) GMP/6.2.1-GCCcore-12.3.0
6) numactl/2.0.16-GCCcore-12.3.0 17) bzip2/1.0.8-GCCcore-12.3.0 28) Java/20.0.2
7) UCX/1.18.1-GCCcore-12.3.0 18) XZ/5.4.2-GCCcore-12.3.0 29) libgit2/1.6.4-GCC-12.3.0
8) UCC/1.3.0-GCCcore-12.3.0 19) PCRE2/10.42-GCCcore-12.3.0 30) nodejs/18.18.2-GCCcore-12.3.0
9) OpenMPI/4.1.5-GCC-12.3.0 20) ncurses/6.4-GCCcore-12.3.0 31) LibTIFF/4.4.0-GCCcore-12.3.0
10) OpenBLAS/0.3.23-GCC-12.3.0 21) libreadline/8.2-GCCcore-12.3.0 32) R/4.3.2-foss-2023a
11) FlexiBLAS/3.3.1-GCC-12.3.0 22) libxml2/2.11.4-GCCcore-12.3.0
Where:
S: Module is Sticky, requires --force to unload or purge
Notice that our initial list of modules has increased by 30. When we loaded R, it also loaded all of it’s dependencies along with all the dependencies of those modules.
Before moving onto the next session lets use module purge again to return to the minimal environment.
10:00:00 login01 $ module purge
The following modules were not unloaded:
(Use "module --force purge" to unload all):
1) NeSI/zen3
Software Versioning
So far, we’ve learned how to load and unload software packages. However, we have not yet addressed the issue of software versioning. At some point or other, you will run into issues where only one particular version of some software will be suitable. Perhaps a key bugfix only happened in a certain version, or version X broke compatibility with a file format you use. In either of these example cases, it helps to be very specific about what software is loaded.
Let’s examine the output of module avail more closely.
10:00:00 login01 $ module avail
-----------------/opt/nesi/CS400_centos7_bdw/modules/all ------------------
fgbio/2.0.2 PyQt/5.10.1-gimkl-2018b-Python-3.7.3
FIGARO/1.1.2-gimkl-2022a-Python-3.11.3 PyQt/5.12.1-gimkl-2020a-Python-3.8.2 (D)
File-Rename/1.13-GCC-9.2.0 Python-Geo/2.7.16-gimkl-2018b
FileSender/2.49.0-foss-2023a Python-Geo/3.7.3-gimkl-2018b
Filtlong/0.2.0 Python-Geo/3.8.2-gimkl-2020a
FimTyper/1.0.1-GCC-11.3.0-Perl-5.34.1 Python-Geo/3.9.5-gimkl-2020a
fineSTRUCTURE/4.1.1-gimkl-2022a-Perl-5.34.1 Python-Geo/3.9.9-gimkl-2020a
flatbuffers/1.12.0-GCCcore-9.2.0 Python-Geo/3.10.5-gimkl-2022a
flex/2.6.4-GCCcore-7.4.0 Python-Geo/3.11.3-gimkl-2022a (D)
FlexiBLAS/3.1.3-GCC-11.3.0 Python/2.7.16-gimkl-2018b
FlexiBLAS/3.3.1-GCC-12.3.0 Python/2.7.18-gimkl-2020a
FlexiBLAS/3.4.4-intel-compilers-2023.2.1 (D) Python/3.7.3-gimkl-2018b
FLTK/1.3.5-GCCcore-9.2.0 Python/3.8.1-gimkl-2018b
Flye/2.4.2-gimkl-2018b-Python-2.7.16 Python/3.8.2-gimkl-2020a
Flye/2.7.1-gimkl-2020a-Python-2.7.18 Python/3.9.5-gimkl-2020a
Flye/2.8.3-gimkl-2020a-Python-3.8.2 Python/3.9.9-gimkl-2020a
Flye/2.9-gimkl-2020a-Python-3.8.2 Python/3.10.5-gimkl-2022a
Flye/2.9.1-gimkl-2022a-Python-3.10.5 Python/3.11.3-gimkl-2022a
Flye/2.9.3-gimkl-2022a-Python-3.11.3 Python/3.11.6-foss-2023a (D)
[removed most of the output here for clarity]
Where:
S: Module is Sticky, requires --force to unload or purge
L: Module is loaded
D: Default Module
If the avail list is too long consider trying:
"module --default avail" or "ml -d av" to just list the default modules.
"module overview" or "ml ov" to display the number of modules for each name.
Use "module spider" to find all possible modules and extensions.
Use "module keyword key1 key2 ..." to search for all possible modules matching any of the "keys".
Let’s take a closer look at the Python modules. There are many applications
that are run using python and may fail to run if the wrong version is loaded.
In this case, there are many different versions: Python/3.7.3-gimkl-2018b,
Python/3.8.1-gimkl-2018b through to the newest versions.
How do we load each copy and which copy is the default?
In this case, Python/3.11.6-foss-2023a has a (D) next to it. This indicates that it is the
default — if we type module load Python, as we did above, this is the copy that will be
loaded.
10:00:00 login01 $ module load Python
10:00:00 login01 $ python3 --version
Python 3.11.6
So how do we load the non-default copy of a software package? In this case, the
only change we need to make is be more specific about the module we are
loading. There are many other Python versions. To load a
non-default module, the only change we need to make to our module load
command is to add the version number after the /.
10:00:00 login01 $ module load Python/3.11.3-gimkl-2022a
The following have been reloaded with a version change:
1) GCC/12.3.0 => GCC/11.3.0
2) GCCcore/12.3.0 => GCCcore/11.3.0
3) HDF5/1.14.3-gompi-2023a => HDF5/1.12.2-gimpi-2022a
4) OpenSSL/1.1 => OpenSSL/1.1.1k-GCCcore-11.3.0
5) Python/3.11.6-foss-2023a => Python/3.11.3-gimkl-2022a
6) SQLite/3.42.0-GCCcore-12.3.0 => SQLite/3.36.0-GCCcore-11.3.0
7) Szip/2.1.1-GCCcore-12.3.0 => Szip/2.1.1-GCCcore-11.3.0
8) Tcl/8.6.10-GCCcore-12.3.0 => Tcl/8.6.10-GCCcore-11.3.0
9) Tk/8.6.10-GCCcore-12.3.0 => Tk/8.6.10-GCCcore-11.3.0
10) UCX/1.18.1-GCCcore-12.3.0 => UCX/1.12.1-GCC-11.3.0
11) XZ/5.4.2-GCCcore-12.3.0 => XZ/5.2.5-GCCcore-11.3.0
12) ZeroMQ/4.3.5-GCCcore-12.3.0 => ZeroMQ/4.3.4-GCCcore-11.3.0
13) binutils/2.40-GCCcore-12.3.0 => binutils/2.38-GCCcore-11.3.0
14) bzip2/1.0.8-GCCcore-12.3.0 => bzip2/1.0.8-GCCcore-11.3.0
15) cURL/8.3.0-GCCcore-12.3.0 => cURL/7.83.1-GCCcore-11.3.0
16) freetype/2.13.2-GCCcore-12.3.0 => freetype/2.11.1-GCCcore-11.3.0
17) libjpeg-turbo/2.1.5.1-GCCcore-12.3.0 => libjpeg-turbo/2.1.3-GCCcore-11.3.0
18) libpng/1.6.40-GCCcore-12.3.0 => libpng/1.6.37-GCCcore-11.3.0
19) libreadline/8.2-GCCcore-12.3.0 => libreadline/8.1-GCCcore-11.3.0
20) libxml2/2.11.4-GCCcore-12.3.0 => libxml2/2.9.10-GCCcore-11.3.0
21) libxslt/1.1.38-GCCcore-12.3.0 => libxslt/1.1.34-GCCcore-11.3.0
22) ncurses/6.4-GCCcore-12.3.0 => ncurses/6.2-GCCcore-11.3.0
23) netCDF/4.9.2-gompi-2023a => netCDF/4.8.1-gimpi-2022a
24) numactl/2.0.16-GCCcore-12.3.0 => numactl/2.0.14-GCC-11.3.0
25) zlib/1.2.13-GCCcore-12.3.0 => zlib/1.2.11-GCCcore-11.3.0
Notice how the module command has swapped out versions of the Python module. And now we test which version we are using:
10:00:00 login01 $ python3 --version
Python 3.11.3
We are now left with only those module required to do our work for this project.
10:00:00 login01 $ module list
Currently Loaded Modules:
1) NeSI/zen3 (S) 17) numactl/2.0.14-GCC-11.3.0 33) libreadline/8.1-GCCcore-11.3.0
2) UCC/1.3.0-GCCcore-12.3.0 18) UCX/1.12.1-GCC-11.3.0 34) libxml2/2.9.10-GCCcore-11.3.0
3) OpenMPI/4.1.5-GCC-12.3.0 19) impi/2021.5.1-GCC-11.3.0 35) libxslt/1.1.34-GCCcore-11.3.0
4) OpenBLAS/0.3.23-GCC-12.3.0 20) AlwaysIntelMKL/1.0 36) LegacySystemLibs/.crypto7 (H)
5) FlexiBLAS/3.3.1-GCC-12.3.0 21) imkl/2022.0.2 37) cURL/7.83.1-GCCcore-11.3.0
6) FFTW/3.3.10-GCC-12.3.0 22) gimpi/2022a 38) netCDF/4.8.1-gimpi-2022a
7) gompi/2023a 23) imkl-FFTW/2022.0.2-gimpi-2022a 39) SQLite/3.36.0-GCCcore-11.3.0
8) FFTW.MPI/3.3.10-gompi-2023a 24) gimkl/2022a 40) METIS/5.1.0-GCC-11.3.0
9) ScaLAPACK/2.2.0-gompi-2023a-fb 25) bzip2/1.0.8-GCCcore-11.3.0 41) GMP/6.2.1-GCCcore-11.3.0
10) foss/2023a 26) XZ/5.2.5-GCCcore-11.3.0 42) MPFR/4.1.0-GCC-11.3.0
11) zstd/1.5.5-GCCcore-12.3.0 27) libpng/1.6.37-GCCcore-11.3.0 43) SuiteSparse/5.13.0-gimkl-2022a
12) GCCcore/11.3.0 28) freetype/2.11.1-GCCcore-11.3.0 44) Tcl/8.6.10-GCCcore-11.3.0
13) zlib/1.2.11-GCCcore-11.3.0 29) Szip/2.1.1-GCCcore-11.3.0 45) Tk/8.6.10-GCCcore-11.3.0
14) binutils/2.38-GCCcore-11.3.0 30) HDF5/1.12.2-gimpi-2022a 46) ZeroMQ/4.3.4-GCCcore-11.3.0
15) GCC/11.3.0 31) libjpeg-turbo/2.1.3-GCCcore-11.3.0 47) OpenSSL/1.1.1k-GCCcore-11.3.0
16) libpmi/2-slurm 32) ncurses/6.2-GCCcore-11.3.0 48) Python/3.11.3-gimkl-2022a
Where:
S: Module is Sticky, requires --force to unload or purge
H: Hidden Module
Key Points
Load software with
module load softwareName.Unload software with
module unloadThe module system handles software versioning and package conflicts for you automatically.
Scheduler Fundamentals
Overview
Teaching: 15 min
Exercises: 10 minQuestions
What is a scheduler and why does a cluster need one?
How do I launch a program to run on a compute node in the cluster?
How do I capture the output of a program that is run on a node in the cluster?
Objectives
Run a simple script on the login node, and through the scheduler.
Use the batch system command line tools to monitor the execution of your job.
Inspect the output and error files of your jobs.
Find the right place to put large datasets on the cluster.
Job Scheduler
An HPC system might have thousands of nodes and thousands of users. How do we decide who gets what and when? How do we ensure that a task is run with the resources it needs? This job is handled by a special piece of software called the scheduler. On an HPC system, the scheduler manages which jobs run where and when.
The following illustration compares these tasks of a job scheduler to a waiter in a restaurant. If you can relate to an instance where you had to wait for a while in a queue to get in to a popular restaurant, then you may now understand why sometimes your job do not start instantly as in your laptop.

The scheduler used in this lesson is Slurm. Although Slurm is not used everywhere, running jobs is quite similar regardless of what software is being used. The exact syntax might change, but the concepts remain the same.
Interactive vs Batch
So far, whenever we have entered a command into our terminals, we have received the response immediately in the same terminal, this is said to be an interactive session.
This is all well for doing small tasks, but what if we want to do several things one after another without without waiting in-between? Or what if we want to repeat a series of command again later?
This is where batch processing becomes useful, this is where instead of entering commands directly to the terminal we write them down in a text file or script. Then, the script can be executed by calling it with bash.
Lets try this now, create and open a new file in your current directory called example_job.sh.
(If you prefer another text editor than nano, feel free to use that), we will put to use some things we have learnt so far.
10:00:00 login01 $ nano example_job.sh
#!/bin/bash -e
module purge
module load R
Rscript sum_matrix.r
echo "Done!"
shebang
shebang or shabang, also referred to as hashbang is the character sequence consisting of the number sign (aka: hash) and exclamation mark (aka: bang):
#!at the beginning of a script. It is used to describe the interpreter that will be used to run the script. In this case we will be using the Bash Shell, which can be found at the path/bin/bash. The job scheduler will give you an error if your script does not start with a shebang.
We can now run this script using
10:00:00 login01 $ bash example_job.sh
The following modules were not unloaded:
(Use "module --force purge" to unload all):
1) NeSI/zen3
Running non-MPI task
Shared Memory Running on 'login01.hpc.nesi.org.nz' with 1 CPU(s)
Summing [ 6.000000e+04 x 4.000000e+04 ] matrix, seed = '0'
1% done...
2% done...
...
98% done...
99% done...
100% done...
(Non-MPI) Sums to -29910.135471
Done!
You will get the output printed to your terminal as if you had just run those commands one after another.
Cancelling Commands
You can kill a currently running task by pressing the keys ctrl + c. If you just want your terminal back, but want the task to continue running you can ‘background’ it by pressing ctrl + v. Note, a backgrounded task is still attached to your terminal session, and will be killed when you close the terminal (if you need to keep running a task after you log out, have a look at tmux).
Scheduled Batch Job
Up until now the scheduler has not been involved, our scripts were run directly on the login node (or Jupyter node).
First lets rename our batch script script to clarify that we intend to run it though the scheduler.
mv example_job.sh example_job.sl
File Extensions
A files extension in this case does not in any way affect how a script is read, it is just another part of the name used to remind users what type of file it is. Some common conventions:
.sh: Shell Script.
.sl: Slurm Script, a script that includes a slurm header and is intended to be submitted to the cluster.
.out: Commonly used to indicate the file contains the stdout of some process.
.err: Same as.outbut for stderr.
In order for the job scheduler to do it’s job we need to provide a bit more information about our script.
This is done by specifying slurm parameters in our batch script. Each of these parameters must be preceded by the special token #SBATCH and placed after the shebang, but before the content of the rest of your script.

These parameters tell SLURM things around how the script should be run, like memory, cores and time required.
All the parameters available can be found by checking man sbatch or on the online slurm documentation.
| --job-name | #SBATCH --job-name=MyJob |
The name that will appear when using squeue or sacct |
| --account | #SBATCH --account=nesi99991 |
The account your core hours will be 'charged' to. |
| --time | #SBATCH --time=DD-HH:MM:SS |
Job max walltime |
| --mem | #SBATCH --mem=1500M |
Memory required per node. |
| --output | #SBATCH --output=%j_output.out |
Path and name of standard output file. |
| --ntasks | #SBATCH --ntasks=2 |
Will start 2 MPI tasks. |
| --cpus-per-task | #SBATCH --cpus-per-task=10 |
Will request 10 CPUs per task. |
Comments
Comments in UNIX shell scripts (denoted by
#) are ignored by the bash interpreter. Why is it that we start our slurm parameters with#if it is going to be ignored?Solution
Commented lines are ignored by the bash interpreter, but they are not ignored by slurm. The
#SBATCHparameters are read by slurm when we submit the job. When the job starts, the bash interpreter will ignore all lines starting with#.This is similar to the shebang mentioned earlier, when you run your script, the system looks at the
#!, then uses the program at the subsequent path to interpret the script, in our case/bin/bash(the program ‘bash’ found in the ‘bin’ directory).
Note that just requesting these resources does not make your job run faster, nor does it necessarily mean that you will consume all of these resources. It only means that these are made available to you. Your job may end up using less memory, or less time, or fewer tasks or nodes, than you have requested, and it will still run.
It’s best if your requests accurately reflect your job’s requirements. We’ll talk more about how to make sure that you’re using resources effectively in a later episode of this lesson.
Now, rather than running our script with bash we submit it to the scheduler using the command sbatch (slurm batch).
10:00:00 login01 $ sbatch example_job.sl
Submitted batch job 360064
And that’s all we need to do to submit a job. Our work is done – now the scheduler takes over and tries to run the job for us.
Checking on Running/Pending Jobs
While the job is waiting
to run, it goes into a list of jobs called the queue. To check on our job’s
status, we check the queue using the command
squeue (slurm queue). We will need to filter to see only our jobs, by including either the flag --user <username> or --me.
10:00:00 login01 $ squeue --me
JOBID USER ACCOUNT NAME CPUS MIN_MEM PARTITI START_TIME TIME_LEFT STATE NODELIST(REASON)
231964 yourUsername nesi99991 example_job.sl 1 300M large N/A 1:00 PENDING (Priority)
We can see many details about our job, most importantly is it’s STATE, the most common states you might see are..
PENDING: The job is waiting in the queue, likely waiting for resources to free up or higher prioroty jobs to run. because other jobs have priority.RUNNING: The job has been sent to a compute node and it is processing our commands.COMPLETED: Your commands completed successfully as far as Slurm can tell (e.g. exit 0).FAILED: (e.g. exit not 0).CANCELLED:TIMEOUT: Your job has running for longer than your--timeand was killed.OUT_OF_MEMORY: Your job tried to use more memory that it is allocated (--mem) and was killed.
Cancelling Jobs
Sometimes we’ll make a mistake and need to cancel a job. This can be done with
the scancel command.
In order to cancel the job, we will first need its ‘JobId’, this can be found in the output of ‘squeue –me’.
10:00:00 login01 $ scancel 231964
A clean return of your command prompt indicates that the request to cancel the job was successful.
Now checking squeue again, the job should be gone.
10:00:00 login01 $ squeue --me
JOBID USER ACCOUNT NAME CPUS MIN_MEM PARTITI START_TIME TIME_LEFT STATE NODELIST(REASON)
(If it isn’t wait a few seconds and try again).
Cancelling multiple jobs
We can also cancel all of our jobs at once using the
-uoption. This will delete all jobs for a specific user (in this case, yourself). Note that you can only delete your own jobs.Try submitting multiple jobs and then cancelling them all.
Solution
First, submit a trio of jobs:
10:00:00 login01 $ sbatch example_job.sl 10:00:00 login01 $ sbatch example_job.sl 10:00:00 login01 $ sbatch example_job.slThen, cancel them all:
10:00:00 login01 $ scancel --user yourUsername
Checking Finished Jobs
There is another command sacct (slurm account) that includes jobs that have finished.
By default sacct only includes jobs submitted by you, so no need to include additional commands at this point.
10:00:00 login01 $ sacct
JobID JobName Alloc Elapsed TotalCPU ReqMem MaxRSS State
--------------- ---------------- ----- ----------- ------------ ------- -------- ----------
31060451 example_job.sl 2 00:00:48 00:33.548 1G CANCELLED
31060451.batch batch 2 00:00:48 00:33.547 102048K CANCELLED
31060451.extern extern 2 00:00:48 00:00:00 0 CANCELLED
Note that despite the fact that we have only run one job, there are three lines shown, this because each job step is also shown.
This can be suppressed using the flag -X.
Where’s the Output?
On the login node, when we ran the bash script, the output was printed to the terminal. Slurm batch job output is typically redirected to a file, by default this will be a file named
slurm-<job-id>.outin the directory where the job was submitted, this can be changed with the slurm parameter--output.
Hint
You can use the manual pages for Slurm utilities to find more about their capabilities. On the command line, these are accessed through the
manutility: runman <program-name>. You can find the same information online by searching > “man". 10:00:00 login01 $ man sbatch
Job environment variables
When Slurm runs a job, it sets a number of environment variables for the job. One of these will let us check what directory our job script was submitted from. The
SLURM_SUBMIT_DIRvariable is set to the directory from which our job was submitted. Using theSLURM_SUBMIT_DIRvariable, modify your job so that it prints out the location from which the job was submitted.Solution
10:00:00 login01 $ nano example_job.sh 10:00:00 login01 $ cat example_job.sh#!/bin/bash -e #SBATCH --time 00:00:30 echo -n "This script is running on " hostname echo "This job was launched in the following directory:" echo ${SLURM_SUBMIT_DIR}
Key Points
The scheduler handles how compute resources are shared between users.
A job is just a shell script.
Request slightly more resources than you will need.
Lunch
Overview
Teaching: min
Exercises: minQuestions
Objectives
Key Points
Using resources effectively
Overview
Teaching: 20 min
Exercises: 20 minQuestions
How can I review past jobs?
How can I use this knowledge to create a more accurate submission script?
Objectives
Understand how to look up job statistics and profile code.
Understand job size implications.
Understand problems and limitations involved in using multiple CPUs.
What Resources?
Last time we submitted a job, we did not specify a number of CPUs, and therefore
we were provided the default of 1.
As a reminder, our slurm script example_job.sl currently looks like this.
#!/bin/bash -e
#SBATCH --job-name my_first_job
#SBATCH --account nesi99991
#SBATCH --mem 300M
#SBATCH --time 00:15:00
module purge
module load R
Rscript sum_matrix.r
echo "Done!"
We will now submit the same job again with more CPUs.
We ask for more CPUs using by adding #SBATCH --cpus-per-task 4 to our script.
In order to help keep track of our different jobs, we can change --job-name to something else e.g. 4cpus.
We will also add #SBATCH --output %x.out. --output allows you to specify the name of the file where the stdout will be sent.
%x is a token standing in for --job-name (saves us renaming the output file if we change the job name),
so in this case the output file will be 4cpus.out.
Your script should now look like this:
#!/bin/bash -e
#SBATCH --job-name 4cpu_job
#SBATCH --account nesi99991
#SBATCH --output %x.out
#SBATCH --mem 300M
#SBATCH --time 00:15:00
#SBATCH --cpus-per-task 4
module purge
module load R
Rscript sum_matrix.r
echo "Done!"
And then submit using sbatch as we did before.
10:00:00 login01 $ sbatch example_job.sl
Submitted batch job 360064
Watch
We can prepend any command with
watchin order to periodically (default 2 seconds) run a command. e.g.watch squeue --mewill give us up to date information on our running jobs. Care should be used when usingwatchas repeatedly running a command can have adverse effects. Exitwatchwith ctrl + c.
Note in squeue, the number under cpus, should be ‘8’.
Checking on our job with sacct.
Oh no!
JobID JobName Partition Account AllocCPUS State ExitCode
------------ ---------- ---------- ---------- ---------- ---------- --------
27323464 my_job large nesi99991 4 OUT_OF_ME+ 0:125
27323464.ba+ batch nesi99991 4 OUT_OF_ME+ 0:125
27323464.ex+ extern nesi99991 4 COMPLETED 0:0
To understand why our job failed, we need to talk about the resources involved.
Understanding the resources you have available and how to use them most efficiently is a vital skill in high performance computing.
Below is a table of common resources and issues you may face if you do not request the correct amount.
| Not enough | Too Much | |
|---|---|---|
| CPU | The job will run more slowly than expected, and so may run out of time and get killed for exceeding its time limit. | The job will wait in the queue for longer. You will be charged for CPUs regardless of whether they are used or not. Your fair share score will fall more. |
| Memory | Your job will fail, probably with an 'OUT OF MEMORY' error, segmentation fault or bus error (may not happen immediately). | The job will wait in the queue for longer. You will be charged for memory regardless of whether it is used or not. Your fair share score will fall more. |
| Walltime | The job will run out of time and be terminated by the scheduler. | The job will wait in the queue for longer. |
Measuring Resource Usage of a Finished Job
Since we have already run a job (successful or otherwise), this is the best source of info we currently have.
If we check the status of our finished job using the sacct command we learned earlier.
10:00:00 login01 $ sacct
JobID JobName Alloc Elapsed TotalCPU ReqMem MaxRSS State
--------------- ---------------- ----- ----------- ------------ ------- -------- ----------
31060451 example_job.sl 2 00:00:48 00:33.548 1G CANCELLED
31060451.batch batch 2 00:00:48 00:33.547 102048K CANCELLED
31060451.extern extern 2 00:00:48 00:00:00 0 CANCELLED
With this information, we may determine a couple of things.
Memory efficiency can be determined by comparing ReqMem (requested memory) with MaxRSS (maximum used memory), MaxRSS is given in KB, so a unit conversion is usually required.
[{Efficiency_{mem} = { MaxRSS \over ReqMem}}]
So for the above example we see that 0.1GB (102048K) of our requested 1GB meaning the memory efficincy was about 10%.
CPU efficiency can be determined by comparing TotalCPU(CPU time), with the maximum possible CPU time. The maximum possible CPU time equal to Alloc (number of allocated CPUs) multiplied by Elapsed (Walltime, actual time passed).
[{Efficiency_{cpu} = { TotalCPU \over {Elapsed \times Alloc}}}]
For the above example 33 seconds of computation was done,
where the maximum possible computation time was 96 seconds (2 CPUs multiplied by 48 seconds), meaning the CPU efficiency was about 35%.
Time Efficiency is simply the Elapsed Time divided by Time Requested.
[{Efficiency_{time} = { Elapsed \over Requested}}]
48 seconcds out of 15 minutes requested give a time efficiency of about 5%
Efficiency Exercise
Calculate for the job shown below,
JobID JobName Alloc Elapsed TotalCPU ReqMem MaxRSS State --------------- ---------------- ----- ----------- ------------ ------- -------- ---------- 37171050 Example-job 8 00:06:03 00:23:04 32G FAILED 37171050.batch batch 8 00:06:03 23:03.999 14082672k FAILED 37171050.extern extern 8 00:06:03 00:00.001 0 COMPLETEDa. CPU efficiency.
b. Memory efficiency.
Solution
a. CPU efficiency is
( 23 / ( 8 * 6 ) ) x 100or around 48%.b. Memory efficiency is
( 14 / 32 ) x 100or around 43%.
For convenience, NeSI has provided the command nn_seff <jobid> to calculate Slurm Efficiency (all NeSI commands start with nn_, for NeSI NIWA).
10:00:00 login01 $ nn_seff <jobid>
Job ID: 27323570
Cluster: mahuika
User/Group: username/username
State: COMPLETED (exit code 0)
Cores: 1
Tasks: 1
Nodes: 1
Job Wall-time: 5.11% 00:00:46 of 00:15:00 time limit
CPU Efficiency: 141.30% 00:01:05 of 00:00:46 core-walltime
Mem Efficiency: 93.31% 233.29 MB of 250.00 MB
Knowing what we do now about job efficiency, lets submit the previous job again but with more appropriate resources.
#!/bin/bash -e
#SBATCH --job-name 4cpu_job
#SBATCH --account nesi99991
#SBATCH --output %x.out
#SBATCH --mem 300M
#SBATCH --time 00:15:00
#SBATCH --cpus-per-task 4
module purge
module load R
Rscript sum_matrix.r
echo "Done!"
10:00:00 login01 $ sbatch example_job.sl
Hopefully we will have better luck with this one!
A quick description of Simultaneous Multithreading - SMT (aka Hyperthreading)
Modern CPU cores have 2 threads of operation that can execute independently of one another. SMT is the technology that allows the 2 threads within one physical core to present as multiple logical cores, sometimes referred to as virtual CPUS (vCPUS).
Note: Hyperthreading is Intel’s marketing name for SMT. Both Intel and AMD CPUs have SMT technology.
Some types of processes can take advantage of multiple threads, and can gain a performance boost. Some software is specifically written as multi-threaded. You will need to check or test if your code can take advantage of threads (we can help with this).
However, because each thread shares resources on the physical core,
there can be conflicts for resources such as onboard cache.
This is why not all processes get a performance boost from SMT and in fact can
run slower. These types of jobs should be run without multithreading. There
is a Slurm parameter for this: --hint=nomultithread
SMT is why you are provided 2 CPUs instead of 1 as we do not allow 2 different jobs to share a core. This also explains why you will sometimes see CPU efficiency above 100%, since CPU efficiency is based on core and not thread.
For more details please see our documentation on Hyperthreading
Measuring the System Load From Currently Running Tasks
On Mahuika, we allow users to connect directly to compute nodes from the login node. This is useful to check on a running job and see how it’s doing, however, we only allow you to connect to nodes on which you have running jobs.
The most reliable way to check current system stats is with htop.
htop is an interactive process viewer that can be launched from command line.
Finding job node
Before we can check on our job, we need to find out where it is running.
We can do this with the command squeue --me, and looking under the ‘NODELIST’ column.
10:00:00 login01 $ squeue --me
JOBID USER ACCOUNT NAME CPUS MIN_MEM PARTITI START_TIME TIME_LEFT STATE NODELIST(REASON)
26763045 cwal219 nesi99991 test 2 512M large May 11 11:35 14:46 RUNNING wbn144
Now that we know the location of the job (wbn189) we can use ssh to run htop on that node.
10:00:00 login01 $ ssh wbn189 -t htop -u $USER
You may get a message:
ECDSA key fingerprint is SHA256:############################################
ECDSA key fingerprint is MD5:9d:############################################
Are you sure you want to continue connecting (yes/no)?
If so, type yes and Enter
You may also need to enter your cluster password.
If you cannot connect, it may be that the job has finished and you have lost permission to ssh to that node.
Reading Htop
You may see something like this,
top - 21:00:19 up 3:07, 1 user, load average: 1.06, 1.05, 0.96
Tasks: 311 total, 1 running, 222 sleeping, 0 stopped, 0 zombie
%Cpu(s): 7.2 us, 3.2 sy, 0.0 ni, 89.0 id, 0.0 wa, 0.2 hi, 0.2 si, 0.0 st
KiB Mem : 16303428 total, 8454704 free, 3194668 used, 4654056 buff/cache
KiB Swap: 8220668 total, 8220668 free, 0 used. 11628168 avail Mem
PID USER PR NI VIRT RES SHR S %CPU %MEM TIME+ COMMAND
1693 jeff 20 0 4270580 346944 171372 S 29.8 2.1 9:31.89 gnome-shell
3140 jeff 20 0 3142044 928972 389716 S 27.5 5.7 13:30.29 Web Content
3057 jeff 20 0 3115900 521368 231288 S 18.9 3.2 10:27.71 firefox
6007 jeff 20 0 813992 112336 75592 S 4.3 0.7 0:28.25 tilix
1742 jeff 20 0 975080 164508 130624 S 2.0 1.0 3:29.83 Xwayland
1 root 20 0 230484 11924 7544 S 0.3 0.1 0:06.08 systemd
68 root 20 0 0 0 0 I 0.3 0.0 0:01.25 kworker/4:1
2913 jeff 20 0 965620 47892 37432 S 0.3 0.3 0:11.76 code
2 root 20 0 0 0 0 S 0.0 0.0 0:00.02 kthreadd
Overview of the most important fields:
PID: What is the numerical id of each process?USER: Who started the process?RES: What is the amount of memory currently being used by a process (in bytes)?%CPU: How much of a CPU is each process using? Values higher than 100 percent indicate that a process is running in parallel.%MEM: What percent of system memory is a process using?TIME+: How much CPU time has a process used so far? Processes using 2 CPUs accumulate time at twice the normal rate.COMMAND: What command was used to launch a process?
To exit press q.
Running this command as is will show us information on tasks running on the login node (where we should not be running resource intensive jobs anyway).
Running Test Jobs
As you may have to run several iterations before you get it right, you should choose your test job carefully. A test job should not run for more than 15 mins. This could involve using a smaller input, coarser parameters or using a subset of the calculations. As well as being quick to run, you want your test job to be quick to start (e.g. get through queue quickly), the best way to ensure this is keep the resources requested (memory, CPUs, time) small. Similar as possible to actual jobs e.g. same functions etc. Use same workflow. (most issues are caused by small issues, typos, missing files etc, your test job is a jood chance to sort out these issues.). Make sure outputs are going somewhere you can see them.
Serial Test
Often a good first test to run, is to execute your job serially e.g. using only 1 CPU. This not only saves you time by being fast to start, but serial jobs can often be easier to debug. If you confirm your job works in its most simple state you can identify problems caused by paralellistaion much more easily.
You generally should ask for 20% to 30% more time and memory than you think the job will use. Testing allows you to become more more precise with your resource requests. We will cover a bit more on running tests in the last lesson.
Efficient way to run tests jobs using debug QOS (Quality of Service)
Before submitting a large job, first submit one as a test to make sure everything works as expected. Often, users discover typos in their submit scripts, incorrect module names or possibly an incorrect pathname after their job has queued for many hours. Be aware that your job is not fully scanned for correctness when you submit the job. While you may get an immediate error if your SBATCH directives are malformed, it is not until the job starts to run that the interpreter starts to process the batch script.
NeSI has an easy way for you to test your job submission. One can employ the debug QOS to get a short, high priority test job. Debug jobs have to run within 15 minutes and cannot use more that 2 nodes. To use debug QOS, add or change the following in your batch submit script
#SBATCH --qos=debug #SBATCH --time=15:00Adding these SBATCH directives will provide your job with the highest priority possible, meaning it should start to run within a few minutes, provided your resource request is not too large.
Initial Resource Requirements
As we have just discussed, the best and most reliable method of determining resource requirements is from testing, but before we run our first test there are a couple of things you can do to start yourself off in the right area.
Read the Documentation
NeSI maintains documentation that does have some guidance on using resources for some software However, as you noticed in the Modules lessons, we have a lot of software. So it is also advised to search the web for others that may have written up guidance for getting the most out of your specific software.
Ask Other Users
If you know someone who has used the software before, they may be able to give you a ballpark figure.
Next Steps
You can use this knowledge to set up the next job with a closer estimate of its load on the system. A good general rule is to ask the scheduler for 30% more time and memory than you expect the job to need.
Key Points
As your task gets larger, so does the potential for inefficiencies.
The smaller your job (time, CPUs, memory, etc), the faster it will schedule.
What is Parallel Computing
Overview
Teaching: 30 min
Exercises: 15 minQuestions
How do we execute a task in parallel?
What benefits arise from parallel execution?
What are the limits of gains from execution in parallel?
What is the difference between implicit and explicit parallelisation.
Objectives
Prepare a job submission script for the parallel executable.
Methods of Parallel Computing
To understand the different types of Parallel Computing we first need to clarify some terms.
CPU: Unit that does the computations.
Task: Like a thread, but multiple tasks do not need to share memory.
Node: A single computer of the cluster. Nodes are made up of CPUs and RAM.
Shared Memory: When multiple CPUs are used within a single task.
Distributed Memory: When multiple tasks are used.
Which methods are available to you is largely dependent on the nature of the problem and software being used.
Shared-Memory (SMP)
Shared-memory multiproccessing divides work among CPUs or threads, all of these threads require access to the same memory.
Often called Multithreading.
This means that all CPUs must be on the same node, most Mahuika nodes have 128 CPUs.
Shared memory parallelism is used in our example script sum_matrix.r.
Number of threads to use is specified by the Slurm option --cpus-per-task.
Distributed-Memory (MPI)
Distributed-memory multiproccessing divides work among tasks, a task may contain multiple CPUs (provided they all share memory, as discussed previously).
Message Passing Interface (MPI) is a communication standard for distributed-memory multiproccessing. While there are other standards, often ‘MPI’ is used synonymously with Distributed parallelism.
Each task has it’s own exclusive memory, tasks can be spread across multiple nodes, communicating via and interconnect. This allows MPI jobs to be much larger than shared memory jobs. It also means that memory requirements are more likely to increase proportionally with CPUs.
Distributed-Memory multiproccessing predates shared-memory multiproccessing, and is more common with classical high performance applications (older computers had one CPU per node).
Number of tasks to use is specified by the Slurm option --ntasks, because the number of tasks ending up on one node is variable you should use --mem-per-cpu rather than --mem (mem per node) to ensure each task has enough.
Using a combination of Shared and Distributed memory is called Hybrid Parallel.
srun
In order to use distrubuted memory parallelism under Slurm, most commands must be prefixed with
srun.For example
srun Rscript sum_matrix.r.We do not need to prefix commands that do not need access to the distrubuted CPUs, ‘echo’, ‘module load’, etc. Some scientific software will have an equivalent built in and may not require
srun, The best place to look for this information is the software user documentation, or the NeSI support docs.
Embarrassingly Parallel
Possible when tasks are independant and no communication between them is required.
e.g. parameter sweeps and Monte Carlo simulations.
Can be thought of less as running a single job in parallel and more about running multiple serial-jobs simultaneously. Often this will involve running the same process on multiple inputs.
Embarrassingly parallel jobs should be able to scale without any loss of efficiency. If this type of parallelisation is an option, it will almost certainly be the best choice.
The best way to run embarrassingly parallel jobs in Slurm is using a Job Array. Job arrays allow one script to be executed many times in a way that is convenient to submit and manage.
A job array can be specified using --array
If you are specifying --output you will probably want to make use of the %a token, so that each output file contains the array ID.
e.g. --output %x_%a.out will create the output <job name>_<array ID>.out.
If you are writing your own code, then this is something you will probably have to specify yourself.
How to Utilise Multiple CPUs
Requesting extra resources through Slurm only means that more resources will be available, it does not guarantee your program will be able to make use of them.
Generally speaking, Parallelism is either implicit where the software figures out everything behind the scenes, or explicit where the software requires extra direction from the user.
Scientific Software
The first step when looking to run particular software should always be to read the documentation. On one end of the scale, some software may claim to make use of multiple cores implicitly, but this should be verified as the methods used to determine available resources are not guaranteed to work.
Some software will require you to specify number of cores (e.g. -n 8 or -np 16), or even type of paralellisation (e.g. -dis or -mpi=intelmpi).
Occasionally your input files may require rewriting/regenerating for every new CPU combintation (e.g. domain based parallelism without automatic partitioning).
Writing Code
Occasionally requesting more CPUs in your Slurm job is all that is required and whatever program you are running will automagically take advantage of the additional resources. However, it’s more likely to require some amount of effort on your behalf.
It is important to determine this before you start requesting more resources through Slurm
If you are writing your own code, some programming languages will have functions that can make use of multiple CPUs without requiring you to changes your code. However, unless that function is where the majority of time is spent, this is unlikely to give you the performance you are looking for.
Python: Multiproccessing (not to be confused with threading which is not really parallel.)
MATLAB: Parpool
Summary
| Name | Other Names | Slurm Option | Pros/cons |
|---|---|---|---|
| Shared Memory Parallelism | Multithreading, Multiproccessing | --cpus-per-task |
does not have to use interconnect/can only use max one node |
| Distrubuted Memory Parallelism | MPI, OpenMPI | --ntasks and add srun before command |
can use any number of nodes/does have to use interconnect |
| Hybrid | --ntasks and --cpus-per-task and add srun before command |
best of both!/can be tricky to set-up and get right | |
| Job Array | --array |
most efficient form of parallelism/not as flexible and requires more manual set-up in Slurm script | |
| General Purpose GPU | --gpus-per-node |
GPUs can accelerate your code A LOT/sometimes GPUs are all busy |
Running a Parallel Job.
Pick one of the method of Paralellism mentioned above, and modify your
example.slscript to use this method.Solution
What does the printout say at the start of your job about number and location of node.
Key Points
Parallel programming allows applications to take advantage of parallel hardware; serial code will not ‘just work.’
There are multiple ways you can run
Afternoon Break
Overview
Teaching: min
Exercises: minQuestions
Objectives
Key Points
Scaling
Overview
Teaching: 10 min
Exercises: 30 minQuestions
How do we go from running a job on a small number of CPUs to a larger one.
Objectives
Understand scaling procedure.
The aim of these tests will be to establish how a jobs requirements change with size (CPUs, inputs) and ultimately figure out the best way to run your jobs. Unfortunately we cannot assume speedup will be linear (e.g. double CPUs won’t usually half runtime, doubling the size of your input data won’t necessarily double runtime) therefore more testing is required. This is called scaling testing.
In order to establish an understanding of the scaling properties we may have to repeat this test several times, giving more resources each iteration.
Scaling Behavior
Amdahl’s Law
Most computational tasks will have a certain amount of work that must be computed serially.

Eventually your performance gains will plateau.
The fraction of the task that can be run in parallel determines the point of this plateau. Code that has no serial components is said to be “embarrassingly parallel”.
It is worth noting that Amdahl’s law assumes all other elements of scaling are happening with 100% efficient, in reality there are additional computational and communication overheads.
Scaling Exercise
- Find your name in the spreadsheet and modify your
example_job.slto request “x”--cpus-per-task. For example#SBATCH --cpus-per-task 10.- Estimate memory requirement based on our previous runs and the cpus requested, memory is specified with the
--memflag, it does not accept decimal values, however you may specify a unit (K|M|G), if no unit is specified it is assumed to beM. For example#SBATCH --mem 1200.- Now submit your job, we will include an extra argument
--acctg-freq 1. By default SLURM records job data every 30 seconds. This means any job running for less than 30 seconds will not have it’s memory use recorded. Submit the job withsbatch --acctg-freq 1 example_job.sl.- Watch the job with
squeue --meorwatch squeue --me.- On completion of job, use
nn_seff <job-id>.- Record the jobs “Elapsed”, “TotalCPU”, and “Memory” values in the spreadsheet. (Hint: They are the first numbers after the percentage efficiency in output of
nn_seff). Make sure you have entered the values in the correct format and there is a tick next to each entry.Solution

Key Points
Start small.
Test one thing at a time (unit tests).
Record everything.